In the previous section we have introduced a purely phenomenological model for spike-time dependent synaptic plasticity which is at least qualitatively in agreement with experimental results. In this section we take a slightly different approach and discuss how the core idea of this model, the learning window, arises from elementary kinetic processes. We start in Section 10.4.1 with a simple mechanistic model and turn then, in Section 10.4.2 to a more detailed model with saturation. A calcium-based model is the topic of Section 10.4.3. All three models give a qualitative explanation for the learning dynamics on the level of individual spikes.
The AND condition in Hebb's postulate suggests that two biochemical components
are involved in the induction of LTP. We do not wish to speculate on the
nature of these components, but simply call them a and b. We assume that
the first component is generated by a chemical reaction chain triggered by
presynaptic spike arrival. In the absence of further input, the concentration
[a] decays with a time constant ![]() back to its
resting level [a] = 0. A simple way to describe this process is
back to its
resting level [a] = 0. A simple way to describe this process is
To generate the synaptic change, another substance b is needed. The production of b is controlled by a second process triggered by postsynaptic spikes,
Hebbian learning needs both `substances' to be present at the same time, thus
Let us now consider the synaptic change caused by a single presynaptic spike
at
tj(f)![]() 0 and a postsynaptic spike a
ti(f) = tj(f) - s. Integration of
Eqs. (10.29) and (10.30) yields
0 and a postsynaptic spike a
ti(f) = tj(f) - s. Integration of
Eqs. (10.29) and (10.30) yields
![\hbox{\hspace{20mm}
\includegraphics[height=50mm]{Figs-ch-Hebbrules/pout.dat.window1.eps} }
\vspace{-18mm}](img1657.gif) |
Equation (10.34) describes the change caused by a single pair of spikes. Given a train of presynaptic input spikes and a set of postsynaptic output spikes, many combinations of firing times (ti(f), tj(f)) exist. Due to the linearity of the learning equation (10.31), the total change is additive, which is consistent with Eq. (10.14).
The combination of two kinetic processes
a and b thus yields an exponential learning
window as in Eq. (10.16) but with
A+ = A-. The learning window either describes LTP (![]() > 0) or LTD
(
> 0) or LTD
(![]() < 0), but not both.
If we want to have an anti-symmetric learning window with
LTP and LTD we need additional processes as detailed below.
< 0), but not both.
If we want to have an anti-symmetric learning window with
LTP and LTD we need additional processes as detailed below.
For a learning window incorporating both LTP and LTD, we need more microscopic variables. Let us suppose that, as before, we have variables [a] and [b] that contribute to LTP according to (10.31), viz.,
We now set
db = 1/![]() and
dc = 1/
and
dc = 1/![]() . In the limit of
. In the limit of
![]()
![]() 0
and
0
and
![]()
![]() 0, we find the asymmetric two-phase learning window
introduced in Eq. (10.16). Weight changes are now instantaneous. A
postsynaptic spike that is triggered after a presynaptic spike arrival reads
out the current value of [a] and induces LTP by an amount
0, we find the asymmetric two-phase learning window
introduced in Eq. (10.16). Weight changes are now instantaneous. A
postsynaptic spike that is triggered after a presynaptic spike arrival reads
out the current value of [a] and induces LTP by an amount
W(tj(f) - ti(f)) = 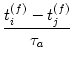 |
(10.37) |
W(tj(f) - ti(f)) = - 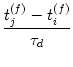 |
(10.38) |
A model for LTP and LTD that is slightly more elaborate than the simplistic
model discussed in the previous section has been developed by
Senn et al. (2001b,1997). This model is based on the assumption that NMDA
receptors can be in one of three different states, a resting state, an `up'
and a `down' state. Transitions between these states are triggered by
presynaptic spike arrival (`rest'![]() `up') and postsynaptic firing
(`rest'
`up') and postsynaptic firing
(`rest'![]() `down'). The actual induction of LTP or LTD, however, requires
another step, namely the activation of so-called second-messengers. The model
assumes two types of second-messenger, one for LTP
and one for LTD. If a presynaptic spike arrives before a postsynaptic
spike,
the upregulation of the NMDA receptors in combination with
the activitation
S1
`down'). The actual induction of LTP or LTD, however, requires
another step, namely the activation of so-called second-messengers. The model
assumes two types of second-messenger, one for LTP
and one for LTD. If a presynaptic spike arrives before a postsynaptic
spike,
the upregulation of the NMDA receptors in combination with
the activitation
S1![]() Sup
of the first second-messenger
triggers
synaptic changes that lead to LTP.
On the other hand, if the presynaptic spike arrives
after postsynaptic firing, the NMDA receptors
are downregulated and the activation
S2
Sup
of the first second-messenger
triggers
synaptic changes that lead to LTP.
On the other hand, if the presynaptic spike arrives
after postsynaptic firing, the NMDA receptors
are downregulated and the activation
S2![]() Sdn
of the other second-messenger triggers LTD;
cf. Fig. 10.9
Sdn
of the other second-messenger triggers LTD;
cf. Fig. 10.9
![\hbox{\hspace{20mm} \includegraphics[width=50mm]{Figs-ch-Hebbrules/Fig13.eps} }](img1672.gif) |
The variables Nup, Ndn, and Nrest describe the portion of NMDA receptors that are in one of the three possible states ( Nup + Ndn + Nrest = 1). In the absence of pre- and postsynaptic spikes, all receptors return to the rest state,
Firing of a postsynaptic spike at time ti(f) leads to a down-regulation of NMDA receptors via
The secondary messenger
Sup, which finally leads to LTP, is
activated by postsynaptic spikes, but only if up-regulated NMDA channels are
available. In the absence of postsynaptic spikes the concentration of second
messengers decays with time constant
![]() . Thus
. Thus
Similarly, the other second messenger Sdn is activated by a presynaptic spike provided that there are receptors in their down regulated state, i.e.,
Long-term potentiation (weight increase) depends on the presence of
Sup,
long-term depression (weight decrease) on
Sdn. This is described by
For low pre- and postsynaptic firing rates, saturation effects can be
neglected and Eq. (10.46) is equivalent to the elementary model
discussed in Section 10.4.1. Let us assume
that a single spike induces a small change (
rdn, rup ![]() 1) so that we can
use
Nrest
1) so that we can
use
Nrest ![]() 1 in Eqs. (10.42) and (10.43). The
equations for
Nup and
Ndn are then identical to those for the
`substances' [a] and [d] in Eqs. (10.29), (10.35),
and (10.36).
1 in Eqs. (10.42) and (10.43). The
equations for
Nup and
Ndn are then identical to those for the
`substances' [a] and [d] in Eqs. (10.29), (10.35),
and (10.36).
Let us furthermore assume that interspike intervals are long compared to the
decay time constants
![]() ,
,![]() in Eqs. (10.44)
and (10.45). Then
Sup is negligible except during and
shortly after a postsynaptic action potential. At the moment of postsynaptic
firing,
Sup `reads out' the current value of
Nup; cf.
Eq. (10.44). If this value is large than
in Eqs. (10.44)
and (10.45). Then
Sup is negligible except during and
shortly after a postsynaptic action potential. At the moment of postsynaptic
firing,
Sup `reads out' the current value of
Nup; cf.
Eq. (10.44). If this value is large than
![]() , it
triggers a positive weight change; cf. Eq. (10.46). Similarly, at the
moment of presynaptic spike arrival
Sdn `reads out' the value of
Ndn and triggers a weight decrease. Thus, in this limit, the model
of Senn et al. corresponds to an exponential time window
, it
triggers a positive weight change; cf. Eq. (10.46). Similarly, at the
moment of presynaptic spike arrival
Sdn `reads out' the value of
Ndn and triggers a weight decrease. Thus, in this limit, the model
of Senn et al. corresponds to an exponential time window
If we assume that all decay time constants are much longer than typical
interspike intervals then the variables
Nup/dn and
Sup/dn will finally reach a steady state. If we neglect
correlations between pre- and postsynaptic neuron by replacing spike trains by
rates, we can solve for these stationary states,
It has been recognized for a long time that calcium ions are an important second messenger for the induction of LTP and LTD in Hippocampus (Malinow et al., 1989; Malenka et al., 1988; Lisman, 1989) and cerebellum (Konnerth and Eilers, 1994; Lev-Ram et al., 1997). Particularly well investigated are `NMDA synapses' in the hippocampus (Dudek and Bear, 1992; Bliss and Collingridge, 1993; Bindman et al., 1991; Collingridge et al., 1983) where calcium ions can enter the cell through channels that are controlled by a glutamate receptor subtype called NMDA (N-methyl-D-aspartic acid) receptor; cf. Section 2.4.2. These channels are involved in transmission of action potentials in glutamatergic (excitatory) synapses. If an action potential arrives at the presynaptic terminal, glutamate, the most common excitatory neurotransmitter, is released into the synaptic cleft and diffuses to the postsynaptic membrane where it binds to NMDA and AMPA receptors. The binding to AMPA receptors results in the opening of the associated ion channels and hence to a depolarization of the postsynaptic membrane. Channels controlled by NMDA receptors, however, are blocked by magnesium ions and do not open unless the membrane is sufficiently depolarized so as to remove the block. Therefore, calcium ions can enter the cell only if glutamate has been released by presynaptic activity and if the postsynaptic membrane is sufficiently depolarized. The calcium influx is the first step in a complex bio-chemical pathway that leads ultimately to a modification of the glutamate-sensitivity of the postsynaptic membrane.
Biophysical models of Hebbian plasticity (Schiegg et al., 1995; Zador et al., 1990; Holmes and Levy, 1990; Shouval et al., 2001; Gold and Bear, 1994; Lisman, 1989) contain two essential components, viz. a description of intracellular calcium dynamics, in particular a model of calcium entry through NMDA synapses; and a hypothesis of how the concentration of intracellular calcium influences the change of synaptic efficacy. In this section we give a simplified account of both components. We start with a model of NMDA synapses and turn then to the so-called calcium control hypothesis of Shouval et al. (2001).
We have emphasized in Sections 10.1-10.3 that all Hebbian learning rules contain a term that depends on the correlation between the firings of pre- and postsynaptic neurons. The signaling chain that leads to a weight change therefore has to contain a nonlinear processing step that requires that pre- and postsynaptic neurons are active within some short time window. Synaptic channels controlled by NMDA receptors are an excellent candidate for a biophysical implementation of this condition of `coincidence' because the opening of the channel requires both the presence of glutamate which reflects presynaptic activity and, in order to remove the magnesium block, a depolarization of the postsynaptic membrane (Mayer et al., 1984; Nowak et al., 1984). A strong depolarization of the postsynaptic membrane does occur, for example, during the back propagation of an action potential into the dendritic tree (Stuart and Sakmann, 1994; Linden, 1999), which is a signature for postsynaptic activity.
![\hbox{{\bf A} \hspace{65mm} {\bf B}} \hbox{\hspace{10mm}
\includegraphics[width...
....eps} \hspace{20mm}
\includegraphics[width=30mm]{Figs-ch-Hebbrules/BPAP.eps} }](img1695.gif) |
In a simple model of NMDA-receptor controlled channels, the calcium current through the channel is described by
The time course of ![]() is taken as a sum of two exponentials with
the time constant of the slow component in the range of 100ms. If there are
several presynaptic spikes within 100ms, calcium accumulates inside the
cell. The change of the intracellular calcium concentration
[Ca2+]
can be described by
is taken as a sum of two exponentials with
the time constant of the slow component in the range of 100ms. If there are
several presynaptic spikes within 100ms, calcium accumulates inside the
cell. The change of the intracellular calcium concentration
[Ca2+]
can be described by
While the dynamics of NMDA synapses is fairly well understood in terms of the biophysical processes that control receptor binding and channel opening, much less is known about the complex signaling chain that is triggered by calcium and finally leads to a regulation of the synaptic efficacy (Lisman, 1989). Instead of a developing a detailed model, we adopt a phenomenological approach and assume that the change of the synaptic efficacy wij is fully determined by the intracellular calcium concentration [Ca2+]; an assumption that has been called `calcium control hypothesis' (Shouval et al., 2001). More specifically, we write the weight change as
![\hbox{{\bf A} \hspace{65mm} {\bf B}} \hbox{\hspace{10mm}
\includegraphics[width...
...ps} \hspace{20mm}
\includegraphics[width=45mm]{Figs-ch-Hebbrules/tau-ca.eps} }](img1700.gif) |
Figure 10.11 shows the graph of the function
![]() ([Ca2+]) as it is used in the model of Shouval et al. (2001). For a calcium
concentration below
([Ca2+]) as it is used in the model of Shouval et al. (2001). For a calcium
concentration below ![]() , the weight assumes a resting value of w0 = 0.5. For calcium concentrations in the range
, the weight assumes a resting value of w0 = 0.5. For calcium concentrations in the range
![]() < [Ca2+] <
< [Ca2+] < ![]() , the weight tends to decrease, for
[Ca2+] >
, the weight tends to decrease, for
[Ca2+] > ![]() it
increases. Qualitatively, the curve
it
increases. Qualitatively, the curve
![]() ([Ca2+]) reproduces
experimental results that suggest that a high level of calcium leads to an
increase whereas an intermediate level of calcium leads to a decrease of
synaptic weights. We will see below that the BCM rule of Eq. (10.12)
is closely related to the function
([Ca2+]) reproduces
experimental results that suggest that a high level of calcium leads to an
increase whereas an intermediate level of calcium leads to a decrease of
synaptic weights. We will see below that the BCM rule of Eq. (10.12)
is closely related to the function
![]() [Ca2+].
[Ca2+].
The time constant
![]() ([Ca2+]) in
the model equation (10.55) decreases rapidly with increasing
calcium concentration; cf. Fig. 10.11B. The specific dependence
has been taken as
([Ca2+]) in
the model equation (10.55) decreases rapidly with increasing
calcium concentration; cf. Fig. 10.11B. The specific dependence
has been taken as
| (10.54) |
In order to complete the definition of the model, we need to introduce a description of the membrane potential ui of the postsynaptic neuron. As in the simple spiking neuron model SRM0 (cf. Chapter 4), the total membrane potential is described as
| ui(t) = |
(10.55) |
| (10.56) |
Given the above components of the model, we can understand intuitively how
calcium influx at NMDA synapses leads to spike-time dependent plasticity. Let
us analyze the behavior by comparing the calcium-based model with the
elementary model of Section 10.4.1; cf.
Eqs. (10.29)-(10.31). Binding of glutamate at NMDA
receptors plays the role of the component a that is triggered by presynaptic
firing; the back propagating action potential plays the role of the component
b that is triggered by postsynaptic firing. As a result of the
depolarization caused by the BPAP, the magnesium block is removed and calcium
ions enter the cell. The calcium influx is proportional to the product of the
NMDA-binding, i.e., the factor ![]() in Eq. (10.52), and the
unblocking, i.e., the factor B(u). Finally, the increase in the calcium
concentration leads to a weight change according to
Eq. (10.55).
in Eq. (10.52), and the
unblocking, i.e., the factor B(u). Finally, the increase in the calcium
concentration leads to a weight change according to
Eq. (10.55).
![\begin{minipage}{0.3\textwidth}
{\bf A}
\par\includegraphics[width=\textwidth]{...
...raphics[width=0.9\textwidth]{Figs-ch-Hebbrules/Shouval-Fig2.eps}
\end{minipage}](img1708.gif) |
A single presynaptic spike (without a simultaneous postsynaptic action
potential) leads to a calcium transient that stays below the induction
threshold ![]() ; cf. Fig. 10.12A. If a postsynaptic
spike occurs 10ms before the presynaptic spike arrival, the calcium
transient has a somewhat larger amplitude that attains a level above
; cf. Fig. 10.12A. If a postsynaptic
spike occurs 10ms before the presynaptic spike arrival, the calcium
transient has a somewhat larger amplitude that attains a level above
![]() . As a consequence, the weight wij is reduced. If, however,
the postsynaptic spike occurs one or a few milliseconds after the
presynaptic one, the calcium transient is much larger. The reason is that the
blocking of the NMDA synapse is removed during the time when the NMDA
receptors are almost completely saturated by glutamate. In this case, the
calcium concentration is well above
. As a consequence, the weight wij is reduced. If, however,
the postsynaptic spike occurs one or a few milliseconds after the
presynaptic one, the calcium transient is much larger. The reason is that the
blocking of the NMDA synapse is removed during the time when the NMDA
receptors are almost completely saturated by glutamate. In this case, the
calcium concentration is well above ![]() so that weights increase.
Since the time constant
so that weights increase.
Since the time constant
![]() ([Ca2+]) is shorter in the regime of
LTP induction than in the regime of LTD induction, the positive weight change
is dominant even though the calcium concentration must necessarily pass
through the regime of LTD in order to reach the threshold
([Ca2+]) is shorter in the regime of
LTP induction than in the regime of LTD induction, the positive weight change
is dominant even though the calcium concentration must necessarily pass
through the regime of LTD in order to reach the threshold ![]() . The
resulting time window of learning is shown in Fig. 10.12B.
It exhibits LTP if the presynaptic spike precedes the postsynaptic one by less
than 40ms. If the order of spiking is inverted LTD occurs. LTD can also be
induced by a sequence of `pre-before-post' if the spike time difference is
larger than about 40ms. The reason is that in this case the removal of the
magnesium block (induced by the BPAP) occurs at a moment when the probability
of glutamate binding is reduced; cf. the factor
. The
resulting time window of learning is shown in Fig. 10.12B.
It exhibits LTP if the presynaptic spike precedes the postsynaptic one by less
than 40ms. If the order of spiking is inverted LTD occurs. LTD can also be
induced by a sequence of `pre-before-post' if the spike time difference is
larger than about 40ms. The reason is that in this case the removal of the
magnesium block (induced by the BPAP) occurs at a moment when the probability
of glutamate binding is reduced; cf. the factor ![]() in
Eq. (10.52). As a consequence less calcium enters the cell -
enough to surpass the threshold
in
Eq. (10.52). As a consequence less calcium enters the cell -
enough to surpass the threshold ![]() , but not sufficient to reach the
threshold
, but not sufficient to reach the
threshold ![]() of LTP. We emphasize that the form of the learning
window is not fixed but depends on the frequency of pre- and postsynaptic
spike firing; cf. Fig. 10.12B.
of LTP. We emphasize that the form of the learning
window is not fixed but depends on the frequency of pre- and postsynaptic
spike firing; cf. Fig. 10.12B.
LTP and LTD can also be induced in the absence of postsynaptic spikes
if the membrane potential of the postsynaptic neuron is clamped to a constant
value. A pure spike-time dependent learning rule defined by a learning window
W(tj(f) - ti(f)) is obviously not a suitable description of such an experiment.
The calcium-based model of Shouval et al. (2001), however, can reproduce
voltage-clamp experiments; cf. Fig. 10.12C. Presynaptic
spike arrival at low frequency (
![]() = 0.5 Hz) is `paired' with a
depolarization of the membrane potential of the postsynaptic neuron to a fixed
value u0. If u0 is below -70mV, no significant weight change occurs.
For -70 mV
< u0 < -50 mV LTD is induced, while for u0 > - 50mV LTP is
triggered. These results are a direct consequence of the removal of the
magnesium block at the NMDA synapses with increasing voltage. The mean
calcium concentration - and hence the asymptotic weight value - is
therefore a monotonously increasing function of u0.
= 0.5 Hz) is `paired' with a
depolarization of the membrane potential of the postsynaptic neuron to a fixed
value u0. If u0 is below -70mV, no significant weight change occurs.
For -70 mV
< u0 < -50 mV LTD is induced, while for u0 > - 50mV LTP is
triggered. These results are a direct consequence of the removal of the
magnesium block at the NMDA synapses with increasing voltage. The mean
calcium concentration - and hence the asymptotic weight value - is
therefore a monotonously increasing function of u0.
Finally, we would like to emphasized the close relation between
Fig. 10.12C and the function ![]() of the BCM learning
rule as illustrated in Fig. 10.5. In a simple rate model, the
postsynaptic firing rate
of the BCM learning
rule as illustrated in Fig. 10.5. In a simple rate model, the
postsynaptic firing rate ![]() is a sigmoidal function of the potential,
i.e.,
is a sigmoidal function of the potential,
i.e.,
![]() = g(ui). Thus, the mapping between the two figures is given by
a non-linear transformation of the horizontal axis.
= g(ui). Thus, the mapping between the two figures is given by
a non-linear transformation of the horizontal axis.
© Cambridge University Press
This book is in copyright. No reproduction of any part
of it may take place without the written permission
of Cambridge University Press.
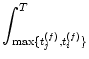 exp
exp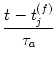 -
- 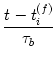
![$\displaystyle \begin{array}{*{2}{c@{\quad}}c} A\,\exp[s/\tau_a] & {\rm for} & s<0 \\ A\,\exp[-s/\tau_b]& {\rm for} & s>0 \, \end{array}$](img1656.gif)
![$\displaystyle \begin{array}{*{2}{c@{\quad}}c} A_+\, \exp[s/\tau_a] - A_-\, \exp...
... A_+\,\exp[-s/\tau_b]- A_- \, \exp[-s/\tau_d] & {\rm for} & s>0 \\ \end{array}$](img1663.gif)
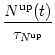 ,
,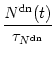 .
.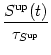 + rS Nup(t) [1 - Sup(t)]
+ rS Nup(t) [1 - Sup(t)] 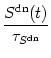 + rS Ndn(t) [1 - Sdn(t)]
+ rS Ndn(t) [1 - Sdn(t)] ![$\displaystyle \begin{array}{*{2}{c@{\qquad}}c} ~A_+(w_{ij})\, \exp[+s/{\tau_{N^...
... A_-(w_{ij}) \, \exp[-s/{\tau_{N^{\rm dn}}}] & {\rm for} & s>0 \\ \end{array}$](img1691.gif)
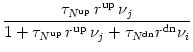
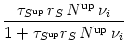
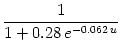
\over \tau_{\rm Ca}}$](img1697.gif) ,
,![$\displaystyle {\Omega([{\rm Ca}^{2+}]) - w_{ij} \over \tau([{\rm Ca}^{2+}])}$](img1699.gif) .
.